![]()
![]()
![]()
![]()
Glycobioinformatics in Influenza Virus
Glycobioinformatics is a synthetic subject based on the rapid development of glycobiology and glycomics in recent decades. The computer technology have played a significant role in the acquisition, storage, analysis, simulation or prediction methods of the data generated by the interaction between glycan and its ligands.
Influenza A viruses (IVs), which belong to the orthomyxoviridae family, consist of eight negative RNA strands. Hemagglutinin (HA) and neuraminidase (NA) are two glycoproteins that are encoded by the IV genome, expressed from segments 4 and 6, respectively. The selection due to various host immune systems and anti-flu drugs accelerate the mutation rates of viral proteins, especially for these two membrane proteins. There are 18 HA subtypes and 11 NA subtypes, designated H1-H18 and N1-N11, respectively. Over 119 combinations of IVs can be isolated from wild birds, which are also the natural reservoir of these viruses (except the H17N10 and H18N11 virus, which, until recently, was isolated only from bat). The species jumping ability of IVs can result in the infections of poultry and mammals, such as chicken, swine, equine or whale species, with different virulence levels. The H1N1, H2N2 and H3N2 viruses have been responsible for tens of millions deaths during the deadly history of human influenza epidemics. Furthermore, the H5N1, H7N7, H7N2, H7N3, H9N2 and H7N9 viruses have been isolated from sporadic human infections and deaths. It is worth noting that the H5N1 virus is the most severe for human and avian species, with sudden onset and high mortality. The mortality rate in hundreds of patients who were hospitalized for H5N1 infections was roughly 59.05%, much higher than the mortality rates of the Spanish Flu or the 2009 influenza pandemic (H1N1).
The existence of N-glycosylation is necessary for viral membrane glycoproteins. The biosynthesis and modification of nascent secretory or membrane proteins occurs in the endoplasmic reticulum and Golgi, N-linked glycans encode crucial information for the folding, maturation, transport or degradation of proteins. To escape both the host’s humoral and cellular immune systems, the potential glycosites in viral envelope proteins can provide the identical glycans as those of the host’s cells to mask the antigenic sites. Additionally, glycosylation also impacts the sensitivity of HA to temperature, the protection of cleavage sites and the stalk domain, and even the receptor-binding preferences. As the ideal model for the influence of N-glycosylation in pathogen-host interaction, the present studies show that the envelope glycoproteins of IVs appear to only have N-glycosylation, with no O-glycosylation and GPI-anchors; hence, the glycosites discussed in this paper only pertain to N-glycosylation site.
Current studies have analyzed the evolutionary dynamics of N-glycosylation sites of select subtypes or HA/NA as a whole. Although most influenza evolution can be accounted for by genetic drift, there is also evidence of adaptive evolution of mutations which are under positive selection. We have extended previous studies of basic similarity alignment scoring of similarity to analyze the position-specific glycosites that are under selective pressure in IVs.Here we present a detailed investigation of the distribution and the evolutionary pattern of the glycosites in the envelope glycoproteins of IVs, especially in the H5N1 virus.
To further explore the binding affinity between HA and SA receptors(SAα2-3Gal or SAα2-6Gal), computational methods were adopted. Commonly, the researchers focused on the interation between the HAs and different SA receptors with less consideration of glycosylation on HA. In the present study, the contribution of glycosylation of HA in IVs for the host recognation was analyzed with the available H5N1 HA sequences and crystal structures. The phylogentic analysis was adopted to invest the key amino-acid residues and glycosylation sites in RBD. Further models construction, molecular dynamics (MD) simulations and docking analysis were utilized to investigate the structure and composition of the glycans for the affection of HA activity.
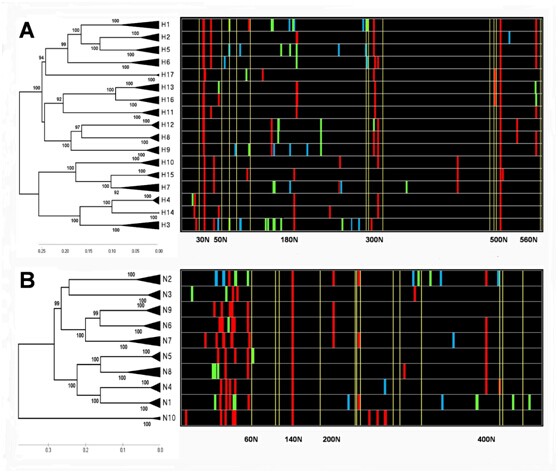
The N-J trees of two glycoproteins in IVs with the corresponding distribution chart of glycosites
The phylogenetic trees of HAs and NAs were constructed using three to ten representative amino acid sequences in each subtype. The distribution charts of glycosites, colored according to the statistics of conservation in each HA or NA subtype (File S2), are shown in various strips. The red, green and blue color represent the levels of conservation of “>95%”, “5%~95%” and “<5%”, respectively. The conserved cysteines are shown in yellow strips. (A) The N-J tree of HA subtypes with the corresponding distribution chart of glycosites. (B) The N-J tree of NA subtypes with the corresponding distribution chart of glycosites.
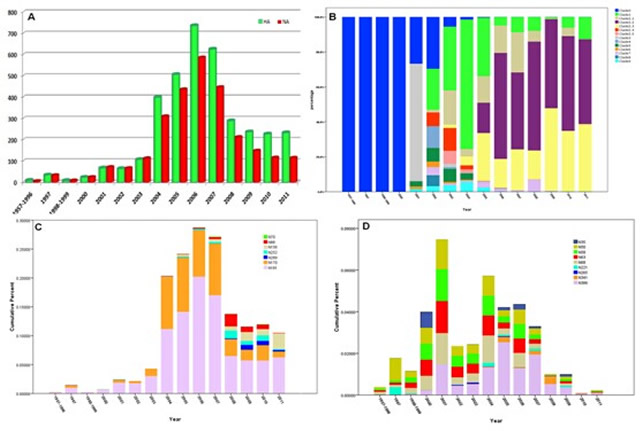
The distribution regularity of glycosites in different H5N1 clades.
The statistical analysis in HA and NA indicated that the glycosites in H5N1 have become more complicated in HA and less influential in NA in the last five years. All the sequences of HA and NA were contained in File S3 and S4. As is shown, the records in 1957~1996, 1998~1999 and 2012 were so limited that should be combined or exclueded. (A) The recording numbers of HA and NA in recent years. The ealiest H5N1 virus record first appeared in 1957 and increased rapidly after 2004. (B) The percentage of various clades or subclades from 1957 to 2011. The early H5N1 viruses belonged to clade 0 and diversified after 2002. The most common clade 2.2 and 2.3 became dominant after 2008. (C) The cumulative percent of unconserved glycosites in HA. The evolution of six unconserved glycosites showed the diversity of N-glycosylation has become common after 2007. (D) The cumulative percent of unconserved glycosites in NA. The evolution of four conserved glycosites in the stalk domain and five unconserved glycosites showed the frequence and diversity have falled rapidly after 2007.
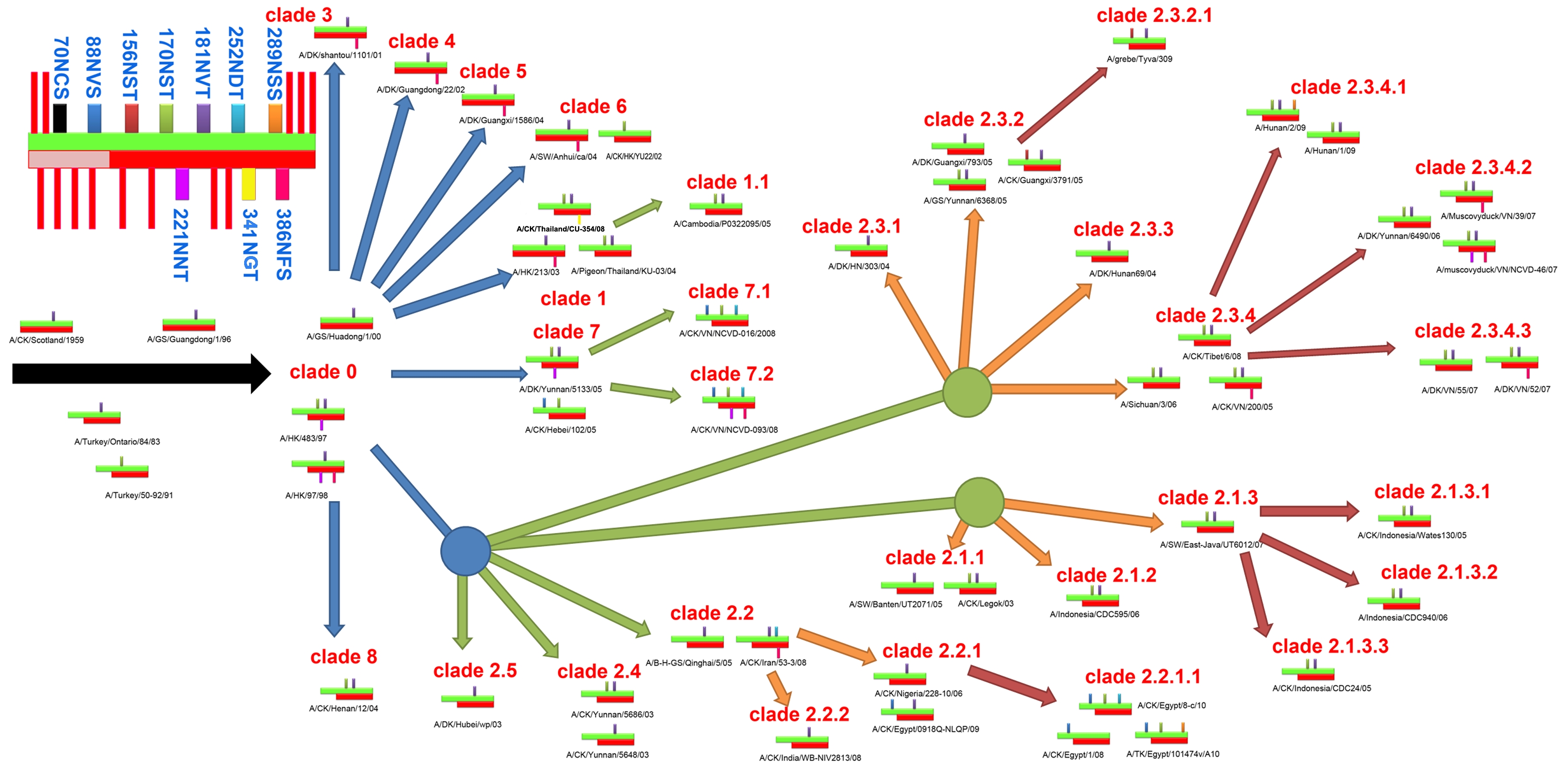
The co-evolution of glycosites between HA and NA in H5N1 virus.
A schematic of diverse glycosites in HA and NA is shown in the top left corner. The green and red crossbands represent HA and NA, respectively, and the lightly colored area in NA is the stem domain. The negligible and highly conserved glycosites are shown in the red strip, while the remainder are labeled in cyan text. All the first-, second-, third- and fourth-order clades are shown with blue, green, orange and crimson arrows, respectively.
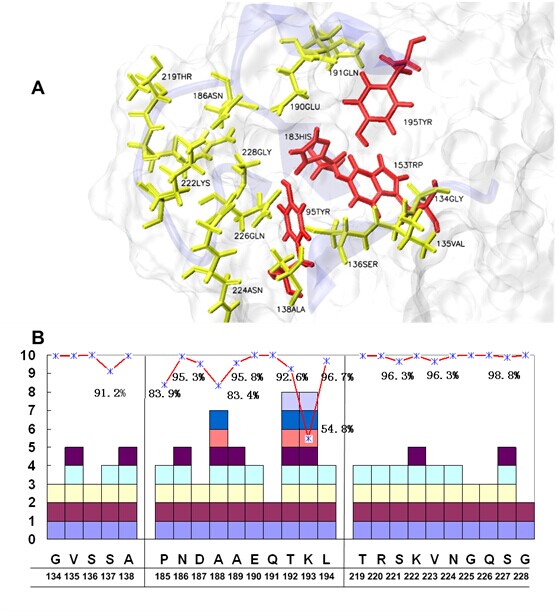
The amino-acid residues in the RBD of H5N1 HA.
(A) The RBD in H5N1 HA consists of three secondary structure elements, Loop130, Loop220 and Helix190, together with four 100% conserved residues at the bottom (red). The residues with a conservation rate greater than 99% are labeled in yellow. (B) The statistics of the conserved amino-acid residues in the RBD. The predominant amino-acid residues are underlined, and the number of different types of mutations are shown as various blocks. Those with conservation rates lower than 99% are marked. All of the residue numbers were adopted from the human H3 numbering system.
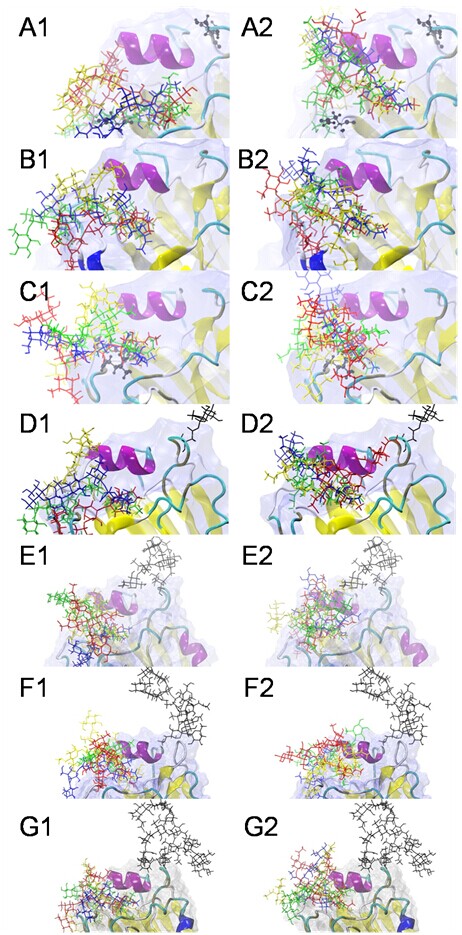
The docking complexes between seven HAs and eight sialoglycans.
SA-a-2,3-Gal receptors are superposed at left, whereas the SAa2,6Gal receptors are superposed at right. LSTa/LSTc are shown in blue, 3DSLNT/6DSLNT are shown in green, BM3/BM6 are shown in red and BG3/BG6 are shown in yellow. The types of receptors deployed distinctive topologies: most SA-a-2,3-Gal receptors were straight and extrorse, whereas the SA-a-2,6-Gal receptors were fishhook-like and ental. (A) 03HK-sialoglycan docking complexes, (B) 04VN-sialoglycan docking complexes, (C) MUT-sialoglycan docking complexes, (D) MG-sialoglycan docking complexes, (E) HM-sialoglycan docking complexes, (F) DS-sialoglycan docking complexes, (G) FG-sialoglycan docking complexes.
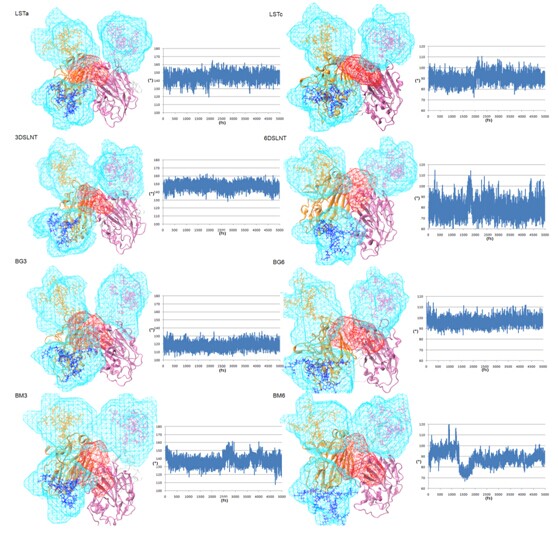
The volumetric topologies of glycans in the Sialoglycans and trimer HA complexes.
As it indicated that the ? angles of sialoglycans are stable during 5ns MD simultion in the trimer HA, all the SA-a-2,3-Gal receptors maintain a straight-like topology (110°<?<180°) and SA-a-2,6-Gal receptors maintain a fishhook-like topology (60°<?<110°) resepectively. The orientations and shapes of the volumetric maps vary dramatically for the N-glycans on 158N and 169N while the sialoglycans varied in the smaller spatial volume in RBD. More complicated N-glycans would sterically hinder the receptor binding, even with different preference.